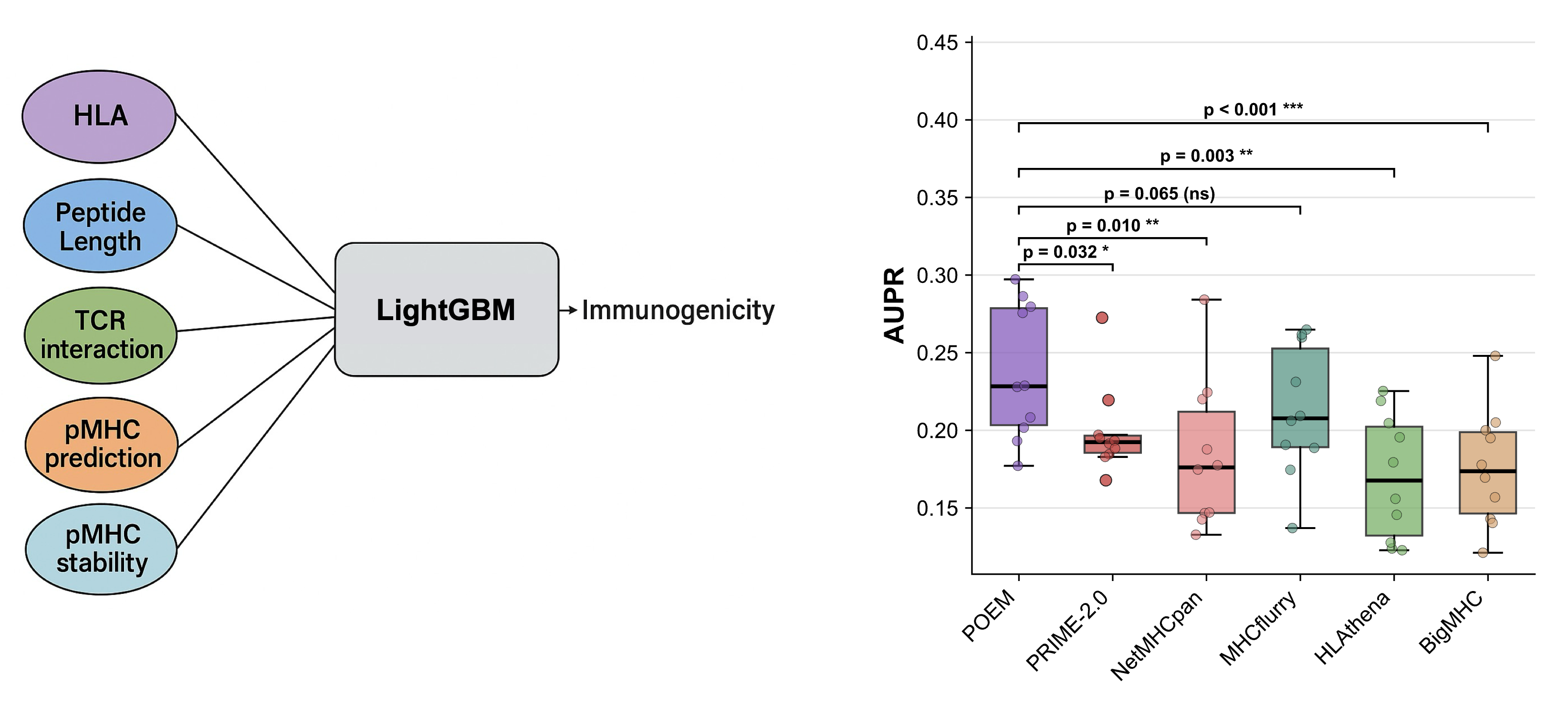
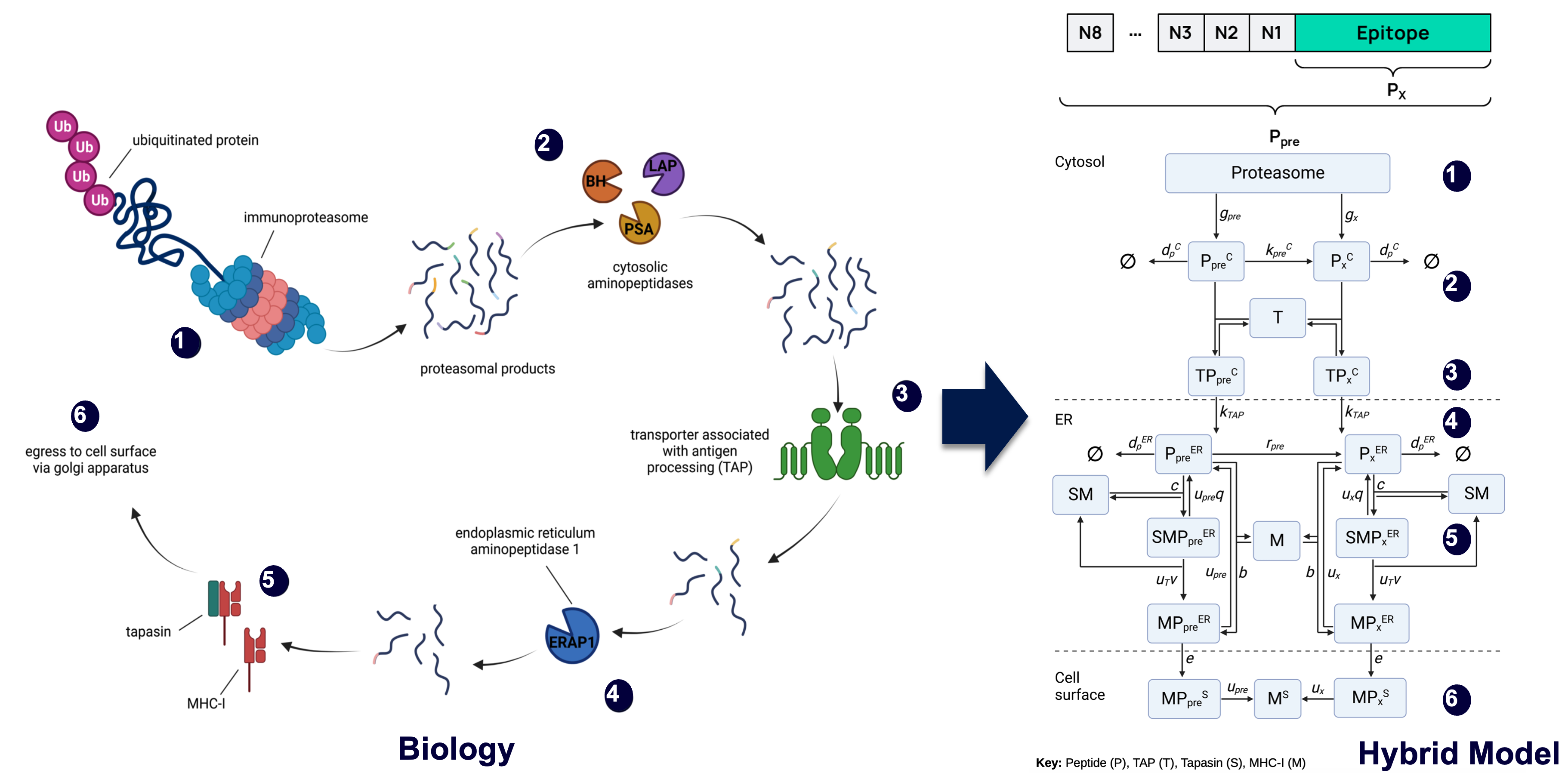
Inference of MHC-I from peptide ligands
Immune cells present peptides on their cell surface in proteins known as MHC-I. The binding affinity and stability between peptides and MHC-I is key to the efficacy of the associated immune response, so predicting these quantaties is desirable. Predictive models are trained using peptide ligands eluted from the surface of cells and identified by mass spectrometry. However, humans have up to 6 different types (alleles) of MHC-I, so it is non-trivial to identify which peptides were bound to each MHC-I allele.
To address this, I fine-tuned a large protein language model (ESM-2) to predict p(MHC-I | peptide). The resulting model could accurately deconvolute multi-allelic mass spectrometry datasets. This work was completed while working at Synteny Biotechnology so is not public.